Proteins
- molecular machines essential to life
- many functions
- consist of chain of amino acids that fold into a 3D structure
- strings over an alphabet of 20 letters
- The exact 3D shape is important for a protein’s function
AlphaFold 3 updates
-
Changes: accommodate more general chemical structures and to improve the data efficiency of learning
-
Within the trunk
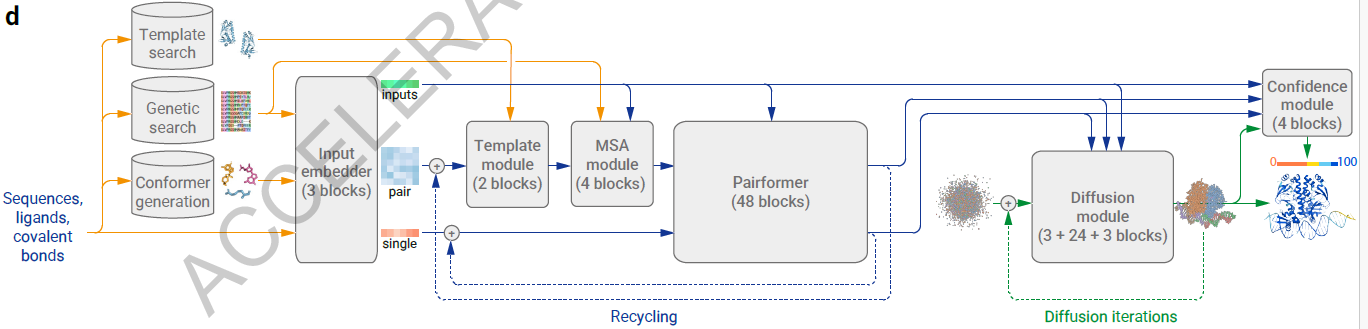
- MSA processing is substantially de-emphasized with a much smaller and simpler MSA embedding block (Supplementary Methods 3.3). Compared to the original Evoformer from AlphaFold 2 the number of blocks are reduced to four, the processing of the MSA representation uses an inexpensive pair-weighted averaging, and only the pair representation is used for later processing steps.
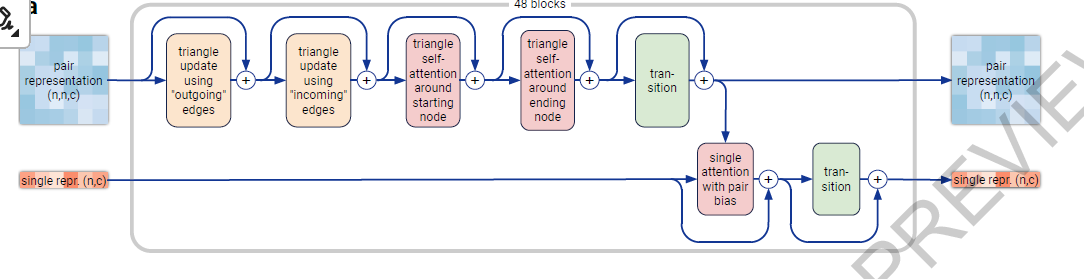
- The “Pairformer” (Fig. 2a, Supplementary Methods 3.6) replaces the “Evoformer” of AlphaFold 2 as the dominant processing block. It operates only on the pair representation and the single representation; the MSA representation is not retained and all information passes via the pair representation. The pair processing and the number of blocks (48) is largely unchanged from AlphaFold 2.
-
DiffusionModule
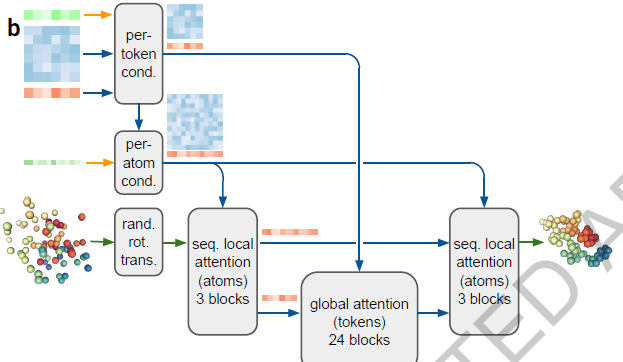
- directly predicts the raw atom coordinates with a Diffusion Module.
- The multiscale nature of the diffusion process (low noise levels induce the network to improve local structure) also allow us to eliminate stereochemical losses and most special handling of bonding patterns in the network, easily accommodating arbitrary chemical components.
Full Form
- Diagram
AlphaFold
- inductive bias of the modules is important
- Physics and geometric insights are built into the network structure
- Inductive bias used:
- Sequence positions de-emphasized (i.e. any amino acids can talk to any amino acid because of folding, so original string is not important)
- Instead, residues that are close in space need to communicate
- Iteratively learn a graph of which are residues are close, while reasoning over this implicit graph as it is being built (could be a soft-graph i.e. an attention matrix)
Inputs
Multiple Sequence Alignment (MSA representation)
- Use previous knowledge of structures
- for the same “function”, the structure should be fairly similar
- thus, given a sequence, they search for “evolutionary” related sequences for which the structure is known to infer a good starting point of the structure (distances)
- Intuition
- Co-evolution, residues in contact must mutate together
- if we see residues mutate, this gives us a prior that they were close
- Stability: if evolution conserves an amino acid at the same spot throughout multiple sequences, it usually means that this amino acid is on the “outside” or “inside” of the protein. e.g. hydrophilic amino acid tend to stay outside
- Co-evolution, residues in contact must mutate together
- How to do it:
- Given a sequence, do a genetics database look-up
- gives us a multiple sequence alignment (MSA) (it’s a stack of sequences)
- Given a sequence, do a genetics database look-up
Residue Pairs representation
- From the input sequence, we create a pairwise representation of all residues
Architecture
Evoformer
Block
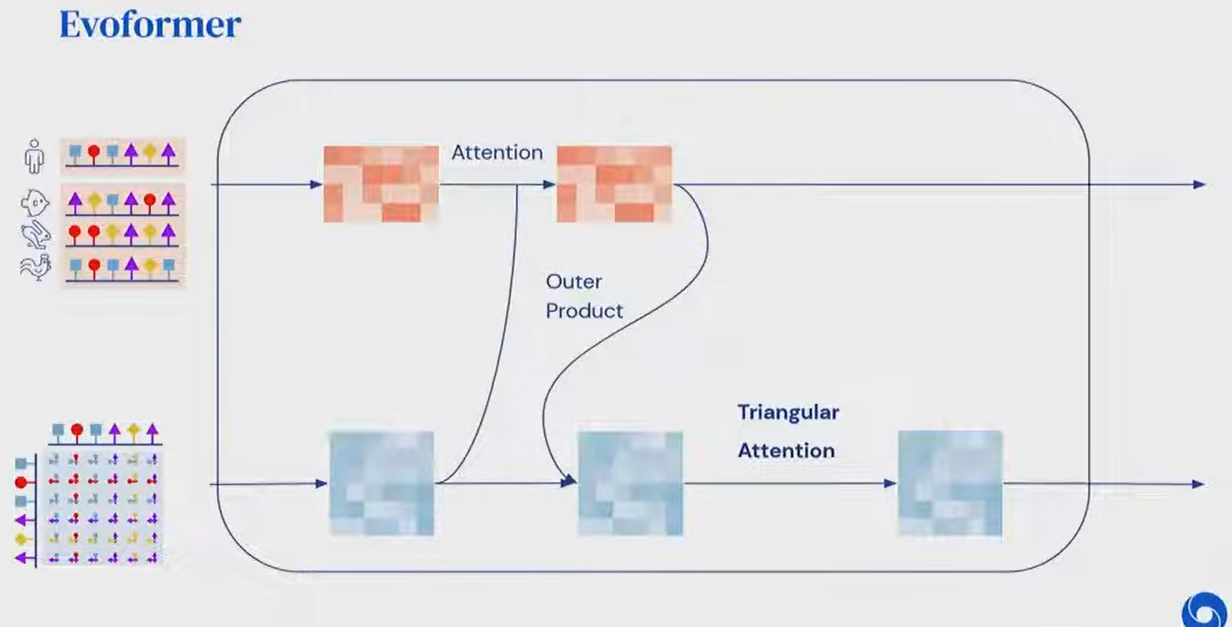
Triangular Attention
- Enforcing Euclidean geometry into the network
- Given 3 points A,B,C
- If distance AB and distance BC known, strong constraint on AC (triangle inequality)
- AC < AB + BC
- Pair Embedding encodes spatial relations
- An update for pair AC should depend on AB and BC
- Given of the pair representation,
- update it by summing (“weighted with attention”) over the row and the column
- different from any-to-any usual attention
Structure Module
- Takes into MSA representation & pair representation ⇒ outputs the 3D structure
- They didn’t the protein backbone as a chain
- They treat as a gas of 3-D rigid bodies (only provided rotation and translation of the body)
- Use a 3-D equivariant transformer architecture
- builds the sides chains form torsion angles
Recycling
- Do 3 forward passes, where the next forward pass is fed the final representations of the previous forward pass (final MSA, final pair representation, structure prediction)
- “Iterative refinement”
Noisy Student Distillation
- Not enough data to do supervised learning
- They iteratively build up a labelled dataset by adding confidently predicted structures from the previously trained model
- Kickstart on PDB data ⇒ iteratively enrich dataset
- They say it acts as a “regularization”
Outputs
Per-residue confidence (pLDDT)
- LDDT = Local Difference Distance Test
- Tells us the per-residue error
- Train a small head to predict this metric during training (and they have labels from PDB)
- Side chains are usually the hardest to be confident on
- Pitfalls
- On a domain scale (rigid body), pLDDT is a good metric
- To assess inter-domain confidence (how big bodies flop around with each other), pLDDT is not good because it is a local metric.
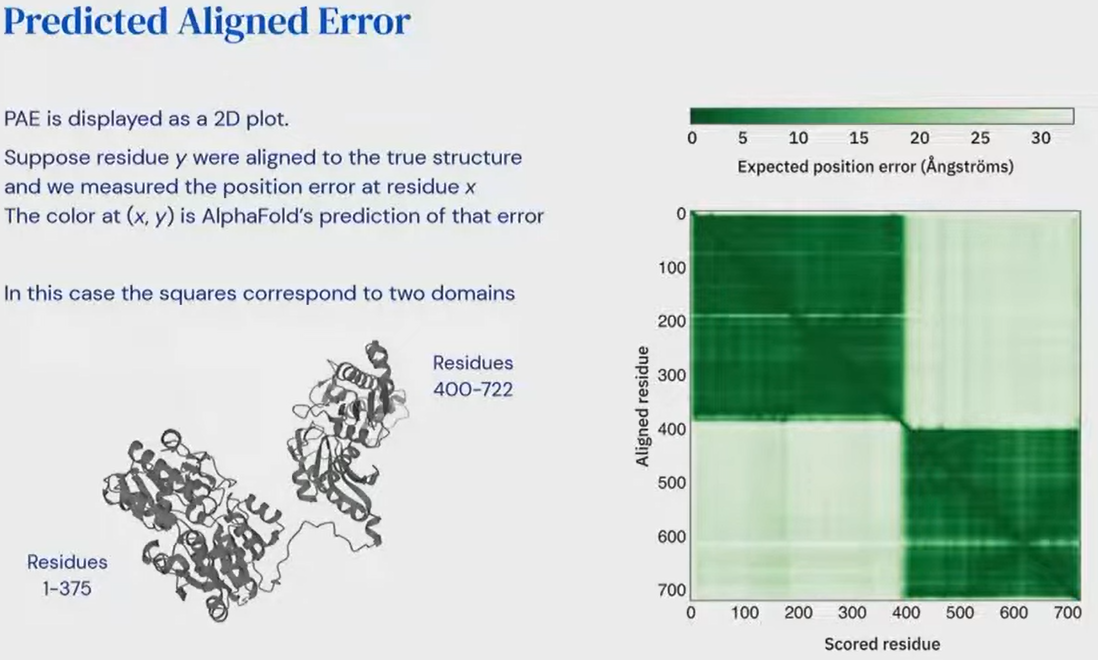
- That’s what “Predicted Aligned Error” is for!
Pairwise confidence (Predicted Aligned Error)
- Gives us a pairwise matrix of errors
- = error for residue if we are aligned to the reference frame of residue .
How AlphaFold understands proteins
- Computational structure prediction is typically underspecified
- context greatly influences structure
- The network implicitly models the missing context
- e.g. presence of zinc atom influencing the side chains